
Tissue remodeling in inflammatory diseases - Translational fibrosis research
Head: Prof. Dr. med. Jörg Distler
The research group investigates the molecular and cellular mechanisms of the interplay between inflammation and tissue response in chronic inflammatory diseases. Using a wide range of different techniques such as human and murine disease models as well as different "omics" approaches in combination with biostatistical methods, we characterize novel molecular mechanisms of pathological tissue remodeling and identify approaches to therapeutically correct these mechanisms. In doing so, we aim to validate our findings in the clinic.
- Subgroups of fibroblasts and macrophages in health and disease.
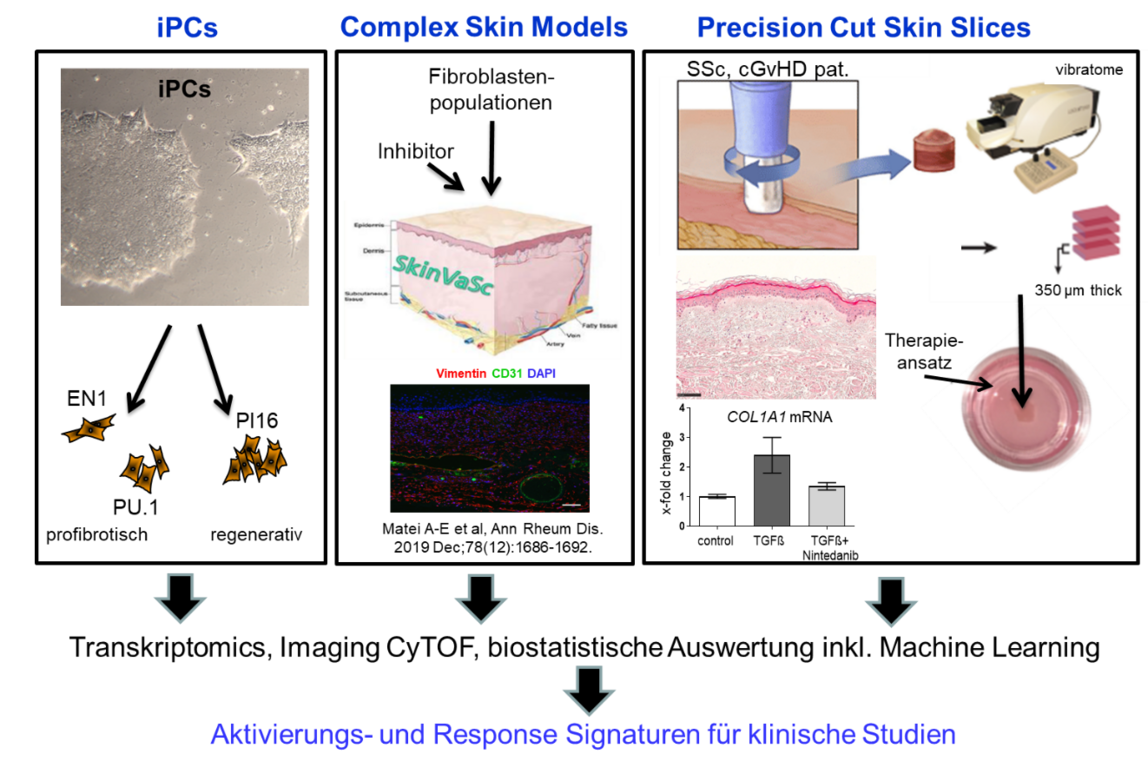
Using combined single cell omics in conjunction with spatial proteomics, we aim to characterize novel fibroblast and macrophage subsets in inflammatory and fibrotic diseases.
Using this approach, we aim to either selectively target pathogenic subpopulations or promote homeostatic (anti-inflammatory and anti-fibrotic) or regenerative subpopulations.
- Epigenetic modifiers are thought to influence pathological tissue memory.
Fibroblasts in fibrotic diseases are endogenously activated and drive fibrosis progression and maintain inflammation, especially in advanced stages of fibrotic diseases. Our goal is to erase the pathological tissue memory and thus break the vicious cycle of epigenetic changes, inflammation and fibroblast activation.
- Interplay between vascular changes, inflammation and tissue fibrosis.
Systemic sclerosis (SSc), as well as other fibrotic diseases, are characterized by early vascular lesions and subsequent inflammation culminating in fibroblast activation. Our studies on the interplay of vascular injury, inflammation and fibroblast activation aim to find new therapeutic approaches to intervene in the earliest stages of pathological tissue remodeling in chronic inflammatory diseases.
- Metabolic alterations and nuclear receptors as potential targets for antifibrotic therapies.
Metabolic changes are proving to be crucial factors in the development of inflammatory and fibrotic diseases. Nuclear receptors are key regulators at the interface of metabolism, inflammation and fibrosis.
Using our extensive repository of preclinical models, specific metabolic assays, and multi-omics, we aim to identify novel metabolic targets for therapeutic intervention in inflammatory diseases with pathological tissue remodeling.
- Novel model systems for fibrotic tissue remodeling.
State-of-the-art preclinical model systems with high predictive value are crucial to minimize the risk of clinical trial failure. We are therefore continuously working with multiple partners to develop new advanced test systems. Our portfolio includes multiple mouse models, primary patient-derived cells, induced pluripotent stem cells, organoids, organ models, organ-on-chip platforms and precisely cut tissue sections.
- From Bench to Bedside
An important goal of our laboratory is the transfer of new results from the laboratory into practice. Therefore, we participate in several clinical trials, biomarker consortia and clinical databases.
| Jörg Distler | Research group leader |
| Debomita Chakraborty | Postdoctoral researcher |
| Andrea-Hermina Györfi | Assistant physician |
| Alexandrú-Emil Matei | Postdoctoral researcher |
| Yun Zhang | Postdoctoral researcher |
| Ariella Zehender | Scientific doctoral student |
| Wolfgang Espach | Medical technical assistant |
| Vladyslav Fedorchenko | Medical technical assistant |
| Aleix-Rius Rigau | Scientific doctoral student |
| Cuong Tran Manh | Scientific doctoral student |
| Xuezhi Hong | Scientific doctoral student |
| Yi-Nan Li | Scientific doctoral student |
| Xiang Zhou | Scientific doctoral student |
Deutsche Forschungsgemeinschaft (DFG)
Individual application, in cooperation with National Natural Science Foundation of China (NSFC), Project partner: Prof. Dr. Hejian Zou "NCOA3 as a coactivator of multiple profibrotic networks in systemic sclerosis." (since 2022)
Individual application "GRK5 as a multimodal regulator of GRK5 TGFβ-dependent fibroblast activation in systemic sclerosis." (since 2021)
Individual application "Characterization of the role of O-GlcNAcylation in osteoclastogenesis." (since 2021)
Individual application "ZAC-1 controls TGFβ-dependent fibroblast activation in fibrotic diseases via regulation of the AP-1 signaling cascade" (since 2020)
CRC 1181 "Liver receptor homolog-1 (LHR-1) regulates inflammation-induced fibrotic tissue remodeling." (2019-2023)
SFB/TRR 221 "Aldehyde dehydrogenase 3A2 (ALDH3A2) as a central regulator of inflammation and tissue fibrosis in scleroderma-form chronic GvHD." (since 2018)
Bundesministerium für Bildung und Forschung (BMBF)
Interdisciplinary research collaborations musculoskeletal diseases:
"MASCARA - Molecular Assessment of Signatures ChAracterizing the Remission of Arthritis” (2020-2022)
Subproject: "Reactions of mesenchymal tissue in rheumatic diseases." (since 2020)
IZKF, Universitätsklinikum Erlangen
A79 - "The nuclear receptor TR4 regulates TGFβ-induced fibroblast activation and tissue fibrosis." (2021-2023)
- Bergmann C*, Distler J*, Treutlein C, Tascilar K, Müller A, Atzinger A, Matei AE, Knitza J, Györfi AH, Lück A, Dees C, Soare A, Ramming A, Schönau V, Distler O, Prante O, Ritt P, Götz TI, Köhner M, Cordes M, Bäuerle T, Kuwert T, Schett G, Schmidkonz C. (2021) 68Ga-FAPI PET/CT for molecular assessment of fibroblast activation and risk evaluation in systemic sclerosis-associated interstitial lung disease: a single-centre, pilot study. Lancet Rheumatology. Volume 3, Issue 3;e185-e194. *contributed equally and corresponding author
This work with Dr. Bergmann translates our preclinical research on fibroblast subsets and mechanisms of fibroblast activation into a clinical application. With this clinical proof-of-concept study, we provide the first evidence that quantification of activated, FAP-expressing fibroblasts by 68Ga-FAPI PET/CT in patients with systemic sclerosis and associated interstitial lung disease allows prognostic stratification into patients with progressive and stable disease. We also demonstrate a correlation between 68Ga-FAPI enrichment and response to antifibrotic therapies. Further studies to evaluate the diagnostic value of 68Ga-FAPI PET/CT in other fibrosing diseases involving other organ systems and in other chronic inflammatory diseases with mesenchymal tissue response are currently underway in collaboration with our nuclear medicine and radiology colleagues and other clinical partners.
- Chakraborty D, Zhu H, Jüngel A, Summa L, Li YN, Matei AE, Zhou X, Huang J, Trinh-Minh T, Chen CW, Lafyatis R, Dees C, Bergmann C, Soare A, Luo H, Ramming A, Schett G, Distler O, Distler JHW. (2020) Fibroblast growth factor receptor 3 activates a network of profibrotic signaling pathways to promote fibrosis in Systemic Sclerosis. Sci Transl Med. 12(563):eaaz5506.
This work demonstrates the selective activation of FGFR3 via its ligand FGF9 in systemic sclerosis and other fibrotic diseases and identifies FGFR3 as a central regulator of several pro-fibrotic networks and many genes that are deregulated in patients. Inhibition of FGFR3/FGF9 corrects the excessive activation of these pro-fibrotic networks and shows potent antifibrotic effects in cultured fibroblasts, in human organoid models, and in several animal models. We are currently negotiating to conduct a proof-of-concept clinical trial with a highly selective FGFR3 inhibitor.
- Dees C, Pötter S, Zhang Y, Bergmann C, Zhou X, Luber M, Wohlfahrt T, Karouzakis E, Ramming A, Gelse K, Yoshimura A, Jaenisch R, Dis tler O, Schett G, Distler JH. (2020) TGFβ-induced epigenetic deregulation of SOCS3 facilitates STAT3-signaling to promote fibrosis. J Clin Invest. 130(5):2347-2363.
We show that TGFβ promotes activation of the pro-fibrotic transcription factor STAT3 through epigenetic inhibition of the endogenous antagonist SOCS3. This mechanism is active in systemic sclerosis with increased DNA methylation of the SOCS3 promoter and decreased SOCS3 expression, resulting in a persistently activated fibroblast phenotype. Inhibition of SOCS3 expression induces a pro-fibrotic phenotype in quiescent fibroblasts, whereas overexpression of SOCS3 or inhibition of TGFβ-regulated DNA methyl transferases corrects the activated phenotype of patient fibroblasts and shows antifibrotic effects in various animal models. Thus, this work identifies DNA methyl transferase inhibitors as a potential therapeutic approach to erase pathological tissue memory.
- Wohlfahrt T, Rauber S, Uebe S, Luber M, Soare A, Ekici A, Weber S, Matei AE, Chen CW, Maier C, Karouzakis E, Kiener HP, Pachera E, Dees C, Beyer C, Daniel C, Gelse K, Kremer AE, Naschberger E, Stürzl M, Butter F, Sticherling M, Finotto S, Kreuter A, Kaplan MH, Jüngel A, Gay S, Nutt SL, Boykin DW, Poon GMK, Distler O, Schett G, Distler JHW, Ramming A. (2019) PU.1 controls fibroblast polarization and tissue fibrosis. Nature. 566(7744):344-349.
In this collaborative project with PD Dr. Ramming, we have identified the transcription factor PU.1 as a central regulator of fibroblast polarization. The expression of PU.1 is repressed in healthy tissue and in inflammatory diseases via different epigenetic mechanisms (histone methylation and miRNAs). This epigenetic control is lost in fibrotic diseases such as systemic sclerosis, liver cirrhosis, or idiopathic pulmonary fibrosis, and PU.1 is expressed by fibroblasts in fibrotic tissues. PU.1 not only stimulates differentiation of quiescent fibroblasts into pro-fibrotic myofibroblasts, but also converts inflammatory fibroblasts into pro-fibrotic fibroblasts when PU.1 expression is forced. Inactivation of PU.1 inhibits multiple pro-fibrotic transcriptional programs and demonstrates antifibrotic effects in models of experimental skin, lung, and liver fibrosis. This work establishes the concept of polarization of fibroblasts into different phenotypes and identifies PU.1 as a key effector of pro-fibrotic polarization.
- Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, Lin NY, Dietel K, Bozec A, Herrmann M, Kaplan MH, Weigmann B, Zaiss MM, Fearon U, Veale DJ, Cañete JD, Distler O, Rivellese F, Pitzalis C, Neurath MF, McKenzie ANJ, Wirtz S, Schett G, Distler JHW, Ramming A. (2017) Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat Med. 23(8):938-944.
In this collaborative project, we show that IL9-induced activation of innate lymphoid cell type 2 (ILC2) plays an important role in anti-inflammation. IL9 induces proliferation of ILC2 and stimulates their potential to activate regulatory T cells. Depletion of IL9 or ILC2 leads to chronicity of inflammatory processes, whereas recombinant IL9 or transfer of ILC2 induces anti-inflammation. Accordingly, the number of ILC2 in patients with rheumatoid arthritis correlates inversely with clinical disease activity.
- Palumbo-Zerr K, Zerr P, Distler A, Fliehr J, Mancuso R, Huang J, Mielenz D, Tomcik M, Fürnrohr BG, Scholtysek C, Dees C, Beyer C, Krönke G, Metzger D, Distler O, Schett G, Distler JH. (2015) Orphan nuclear receptor NR4A1 regulates transforming growth factor-β signaling and fibrosis. Nat Med. 21(2):150-8.
In this project, we characterize the nuclear receptor NR4A1 as a novel feedback mechanism within the TGFβ signaling cascade. We show that NR4A1 is induced in a TGFβ-dependent manner in physiological tissue responses such as normal wound healing and then inhibits the expression of TGFβ target genes by recruiting a repressor complex. In fibrotic disease, this feedback inhibition is abrogated by epigenetic and posttranslational mechanisms, NR4A1 is not induced, and pro-fibrotic target genes are continuously expressed. Treatment with NR4A1 agonists can restore this feedback inhibition and shows antifibrotic effects in mouse models of skin, lung, kidney, and liver fibrosis. Here, we identify for the first time the inactivation of a homeostatic molecular feedback inhibition mechanism as the cause of the chronic tissue response in fibrotic diseases. In collaboration with various pharmaceutical companies, we are working on optimized NR4A1 agonists for use in clinical trials in fibrotic diseases.
- Dees C, Akhmetshina A, Zerr P, Reich N, Palumbo K, Horn A, Jüngel A, Beyer C, Krönke G, Zwerina J, Reiter R, Alenina N, Maroteaux L, Gay S, Schett G, Distler O, Distler JH. (2011) Platelet-derived serotonin links vascular disease and tissue fibrosis. J Exp Med. 208(5):961-72.
This project demonstrated for the first time that platelets play an important role at the interface between vasculopathy and fibrotic tissue remodeling in systemic sclerosis. Platelets are increasingly activated by angiopathy in systemic sclerosis and release serotonin. At the same time, a serotonin receptor, 5HTR2B, is increasingly expressed by fibroblasts in fibrotic skin. Serotonin selectively activates the expression of profibrotic genes via 5HTR2B. Mice with therapeutic inhibition of platelet activation, deficiency of serotonin in platelets, or pharmaceutical or genetic inactivation of 5HTR2B are protected from fibrosis. This work was the starting point for a successful PoC clinical trial with a 5HTR2B antagonist, a planned phase 3 trial, and is the basis for further clinical programs with highly selective 5HTR2B inhibitors in systemic sclerosis and other fibrosing diseases.
- Distler JH, Jüngel A, Huber LC, Seemayer CA, Reich CF 3rd, Gay RE, Michel BA, Fontana A, Gay S, Pisetsky DS, Distler O. (2005) The induction of matrix metalloproteinase and cytokine expression in synovial fibroblasts stimulated with immune cell microparticles. Proc Natl Acad Sci U S A. 102(8):2892-7.
In previous work, we had shown that various immune cells detach microparticles from the cell membrane after activation or upon apoptosis, which are vesicles filled with various intracellular components. In this work, we show that microparticles act as a novel communication mechanism between immune cells and mesenchymal cells during inflammatory processes. Microparticles released by immune cells in the joint induce a proinflammatory phenotype in synovial fibroblasts with release of cytokines and matrix-degrading enzymes. In follow-up work, we also demonstrated that microparticles also regulate angiogenesis through the release of angiogenic chemokines and through effects on endothelial progenitor cells. In addition, we demonstrated that microparticles in plasma correlate with the activity and clinical manifestations of various rheumatologic diseases.
PubMed publication list of Prof. Dr. Jörg Distler