
Gewebeumbau bei entzündlichen Erkrankungen - Translationale Fibroseforschung
Leitung: Prof. Dr. med. Jörg Distler
Die Arbeitsgruppe untersucht die molekularen und zellulären Mechanismen des Wechselspiels zwischen Entzündung und Gewebeantwort in chronisch entzündlichen Erkrankungen. Unter Verwendung einer breiten Palette an verschiedenen Techniken wie humanen und murinen Krankheitsmodellen sowie verschiedenen “omics”-Ansätzen in Kombination mit biostatistischen Verfahren charakterisieren wir neue molekulare Mechanismen des pathologischen Gewebeumbaus und identifizieren Ansätze, um diese Mechanismen therapeutisch zu korrigieren. Dabei zielen wir auf die Validierung unserer Ergebnisse in der Klinik.
- Untergruppen von Fibroblasten und Makrophagen in Gesundheit und Krankheit
Mit Hilfe der kombinierten Einzelzell-Omik in Verbindung mit der räumlichen Proteomik wollen wir neue Fibroblasten- und Makrophagen-Untergruppen bei entzündlichen und fibrotischen Erkrankungen charakterisieren.
Mit diesem Ansatz wollen wir entweder selektiv auf pathogene Subpopulationen abzielen oder homöostatische (entzündungshemmende und antifibrotische) oder regenerative Subpopulationen fördern.
- Epigenetische Modifikatoren sollen das pathologische Gewebegedächtnis beeinflussen
Fibroblasten in fibrotischen Erkrankungen werden endogen aktiviert und treiben das Fortschreiten der Fibrose voran und halten die Entzündung aufrecht, insbesondere in fortgeschrittenen Stadien fibrotischer Erkrankungen. Unser Ziel ist es, das pathologische Gewebegedächtnis zu löschen und so den Teufelskreis aus epigenetischen Veränderungen, Entzündung und Fibroblastenaktivierung zu durchbrechen.
- Wechselspiel zwischen vaskulären Veränderungen, Entzündung und Gewebefibrose
Die Systemische Sklerose (SSc), aber auch andere fibrotische Erkrankungen, sind durch frühe vaskuläre Läsionen und anschließende Entzündungen gekennzeichnet, die in der Aktivierung von Fibroblasten gipfeln. Unsere Studien über das Zusammenspiel von Gefäßverletzungen, Entzündungen und Fibroblastenaktivierung zielen darauf ab, neue therapeutische Ansätze zu finden, um in die frühesten Stadien des pathologischen Gewebeumbaus bei chronischen Entzündungskrankheiten einzugreifen.
- Stoffwechselveränderungen und Kernrezeptoren als potenzielle Ziele für antifibrotische Therapien
Stoffwechselveränderungen erweisen sich als entscheidende Faktoren bei der Entstehung von entzündlichen und fibrotischen Erkrankungen. Kernrezeptoren sind wichtige Regulatoren an der Schnittstelle von Stoffwechsel, Entzündung und Fibrose.
Mit Hilfe unseres umfangreichen Bestands an präklinischen Modellen, spezifischen Stoffwechseluntersuchungen und Multi-omics wollen wir neue Stoffwechselziele für therapeutische Interventionen bei entzündlichen Erkrankungen mit pathologischem Gewebeumbau identifizieren.
- Neuartige Modellsysteme für den Umbau von fibrotischem Gewebe
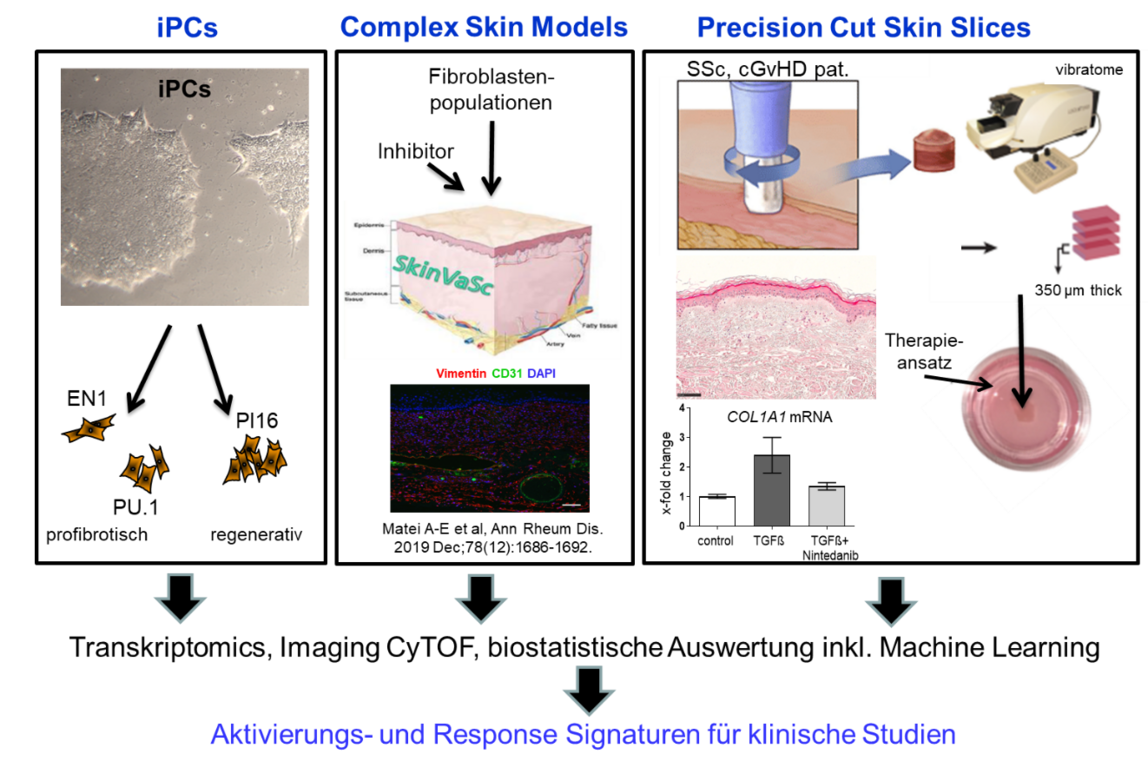
Modernste präklinische Modellsysteme mit hohem Vorhersagewert sind entscheidend, um das Risiko des Scheiterns klinischer Studien zu minimieren. Wir arbeiten daher kontinuierlich mit mehreren Partnern an der Entwicklung neuer, fortschrittlicher Testsysteme. Unser Portfolio umfasst mehrere Mausmodelle, primäre patienteneigene Zellen, induzierte pluripotente Stammzellen, Organoide, Organmodelle, Organ-on-Chip-Plattformen und präzise geschnittene Gewebeschnitte.
- From Bench to Bedside
Ein wichtiges Ziel unseres Labors ist der Transfer neuer Ergebnisse vom Labor in die Praxis. Daher beteiligen wir uns an mehreren klinischen Studien, Biomarker-Konsortien und klinischen Datenbanken.
| Jörg Distler | Arbeitsgruppenleiter |
| Debomita Chakraborty | Postdoktorandin |
| Andrea-Hermina Györfi | Assistenzärztin |
| Alexandrú-Emil Matei | Postdoktorand |
| Yun Zhang | Postdoktorand |
| Ariella Zehender | naturwissenschaftliche Doktorandin |
| Wolfgang Espach | Medizinisch-technischer Assistent |
| Vladyslav Fedorchenko | Medizinisch-technischer Assistent |
| Aleix-Rius Rigau | naturwissenschaftlicher Doktorand |
| Cuong Tran Manh | naturwissenschaftlicher Doktorand |
| Xuezhi Hong | naturwissenschaftlicher Doktorand |
| Yi-Nan Li | naturwissenschaftlicher Doktorand |
| Xiang Zhou | naturwissenschaftlicher Doktorand |
Deutsche Forschungsgemeinschaft (DFG)
Einzelantrag, in Kooperation mit National Natural Science Foundation of China (NSFC), Projektpartner: Prof. Dr. Hejian Zou "NCOA3 als Coaktivator multipler profibrotischer Netzwerke in der systemischen Sklerose" (seit 2022)
Einzelantrag "GRK5 als multimodaler Regulator der GRK5 TGFβ-abhängigen Fibroblastenaktivierung in der systemischen Sklerose" (seit 2021)
Einzelantrag "Charakterisierung der Rolle der O-GlcNAcylierung in der Osteoklastogenese" (seit 2021)
Einzelantrag "ZAC-1 kontrolliert die TGFβ-abhängige Fibroblastenaktivierung in fibrotischen Erkrankungen über Regulation der AP-1 Signalkaskade" (seit 2020)
SFB 1181 "Liver receptor homolog-1 (LHR-1) regulates inflammation-induced fibrotic tissue remodeling." (2019-2023)
SFB/TRR 221 "Aldehyddehydrogenase 3A2 (ALDH3A2) als zentraler Regulator von Entzündungen und Gewebefibrose in der sklerodermieformen chronischen GvHD" (seit 2018)
Bundesministerium für Bildung und Forschung (BMBF)
Interdisziplinäre Forschungsverbünde muskuloskelettale Erkrankungen
"MASCARA - Molekulare Charakterisierung der Remission von Arthritis”
Teilprojekt: "Reaktionen des mesenchymalen Gewebes bei rheumatischen Erkrankungen" (2020-2022)
IZKF, Universitätsklinikum Erlangen
A79 - "Der Kernrezeptor TR4 reguliert die TGFβ-induzierte Fibroblastenaktivierung und Gewebefibrose" (2021-2023)
- Bergmann C*, Distler J*, Treutlein C, Tascilar K, Müller A, Atzinger A, Matei AE, Knitza J, Györfi AH, Lück A, Dees C, Soare A, Ramming A, Schönau V, Distler O, Prante O, Ritt P, Götz TI, Köhner M, Cordes M, Bäuerle T, Kuwert T, Schett G, Schmidkonz C. (2021) 68Ga-FAPI PET/CT for molecular assessment of fibroblast activation and risk evaluation in systemic sclerosis-associated interstitial lung disease: a single-centre, pilot study. Lancet Rheumatology. Volume 3, Issue 3;e185-e194. *contributed equally and corresponding author
Diese Arbeit mit Dr. Bergmann überträgt unsere präklinische Forschung über Fibroblastenuntergruppen und Mechanismen der Fibroblastenaktivierung in eine klinische Anwendung. Mit dieser klinischen Proof-of-Concept-Studie erbringen wir den ersten Nachweis, dass die Quantifizierung von aktivierten, FAP-exprimierenden Fibroblasten durch 68Ga-FAPI PET/CT bei Patienten mit systemischer Sklerose und damit verbundener interstitieller Lungenerkrankung eine prognostische Stratifizierung in Patienten mit fortschreitender und stabiler Erkrankung ermöglicht. Wir zeigen auch eine Korrelation zwischen der 68Ga-FAPI Anreicherung und dem Ansprechen auf antifibrotische Therapien. Weitere Studien zur Bewertung des diagnostischen Werts der 68Ga-FAPI PET/CT bei anderen fibrosierenden Erkrankungen mit Beteiligung anderer Organsysteme und bei anderen chronisch entzündlichen Erkrankungen mit mesenchymaler Gewebereaktion werden derzeit in Zusammenarbeit mit unseren nuklearmedizinischen und radiologischen Kollegen sowie anderen klinischen Partnern durchgeführt.
- Chakraborty D, Zhu H, Jüngel A, Summa L, Li YN, Matei AE, Zhou X, Huang J, Trinh-Minh T, Chen CW, Lafyatis R, Dees C, Bergmann C, Soare A, Luo H, Ramming A, Schett G, Distler O, Distler JHW. (2020) Fibroblast growth factor receptor 3 activates a network of profibrotic signaling pathways to promote fibrosis in Systemic Sclerosis. Sci Transl Med. 12(563):eaaz5506.
Diese Arbeit zeigt die selektive Aktivierung von FGFR3 über seinen Liganden FGF9 bei systemischer Sklerose und anderen fibrosierenden Erkrankungen und identifiziert FGFR3 als zentralen Regulator mehrerer pro-fibrotischer Netzwerke und vieler Gene, die bei Patienten dereguliert sind. Die Hemmung von FGFR3/FGF9 korrigiert die übermäßige Aktivierung dieser pro-fibrotischen Netzwerke und zeigt starke antifibrotische Effekte bei kultivierten Fibroblasten, in menschlichen Organoidmodellen und in verschiedenen Tiermodellen. Wir verhandeln derzeit über die Durchführung einer klinischen Proof-of-Concept-Studie mit einem hochselektiven FGFR3-Inhibitor.
- Dees C, Pötter S, Zhang Y, Bergmann C, Zhou X, Luber M, Wohlfahrt T, Karouzakis E, Ramming A, Gelse K, Yoshimura A, Jaenisch R, Dis tler O, Schett G, Distler JH. (2020) TGFβ-induced epigenetic deregulation of SOCS3 facilitates STAT3-signaling to promote fibrosis. J Clin Invest. 130(5):2347-2363.
Wir zeigen, dass TGFβ die Aktivierung des pro-fibrotischen Transkriptionsfaktors STAT3 durch epigenetische Hemmung des endogenen Antagonisten SOCS3 fördert. Dieser Mechanismus ist bei systemischer Sklerose mit erhöhter DNA-Methylierung des SOCS3-Promotors und verringerter SOCS3-Expression aktiv, was zu einem anhaltend aktivierten Fibroblasten-Phänotyp führt. Die Hemmung der SOCS3-Expression induziert einen pro-fibrotischen Phänotyp in ruhenden Fibroblasten, während die Überexpression von SOCS3 oder die Hemmung von TGFβ-regulierten DNA-Methyl-Transferasen den aktivierten Phänotyp von Patientenfibroblasten korrigiert und in verschiedenen Tiermodellen antifibrotische Effekte zeigt. Somit identifiziert diese Arbeit DNA-Methyl-Transferase-Inhibitoren als einen potenziellen therapeutischen Ansatz, um pathologisches Gewebegedächtnis zu löschen.
- Wohlfahrt T, Rauber S, Uebe S, Luber M, Soare A, Ekici A, Weber S, Matei AE, Chen CW, Maier C, Karouzakis E, Kiener HP, Pachera E, Dees C, Beyer C, Daniel C, Gelse K, Kremer AE, Naschberger E, Stürzl M, Butter F, Sticherling M, Finotto S, Kreuter A, Kaplan MH, Jüngel A, Gay S, Nutt SL, Boykin DW, Poon GMK, Distler O, Schett G, Distler JHW, Ramming A. (2019) PU.1 controls fibroblast polarization and tissue fibrosis. Nature. 566(7744):344-349.
In diesem Kooperationsprojekt mit PD Dr. Ramming haben wir den Transkriptionsfaktor PU.1 als zentralen Regulator der Fibroblastenpolarisation identifiziert. Die Expression von PU.1 wird in gesundem Gewebe und bei entzündlichen Erkrankungen über verschiedene epigenetische Mechanismen (Histon-Methylierung und miRNAs) unterdrückt. Diese epigenetische Kontrolle geht bei fibrotischen Erkrankungen wie systemischer Sklerose, Leberzirrhose oder idiopathischer Lungenfibrose verloren, und PU.1 wird von Fibroblasten in fibrotischen Geweben exprimiert. PU.1 stimuliert nicht nur die Differenzierung von ruhenden Fibroblasten in pro-fibrotische Myofibroblasten, sondern wandelt auch entzündliche Fibroblasten in pro-fibrotische Fibroblasten um, wenn die PU.1-Expression erzwungen wird. Die Inaktivierung von PU.1 hemmt mehrere pro-fibrotische Transkriptionsprogramme und zeigt antifibrotische Wirkungen in Modellen der experimentellen Haut-, Lungen- und Leberfibrose. Diese Arbeit etabliert das Konzept der Polarisierung von Fibroblasten in verschiedene Phänotypen und identifiziert PU.1 als einen zentralen Effektor der pro-fibrotischen Polarisierung.
- Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, Lin NY, Dietel K, Bozec A, Herrmann M, Kaplan MH, Weigmann B, Zaiss MM, Fearon U, Veale DJ, Cañete JD, Distler O, Rivellese F, Pitzalis C, Neurath MF, McKenzie ANJ, Wirtz S, Schett G, Distler JHW, Ramming A. (2017) Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells. Nat Med. 23(8):938-944.
In diesem Gemeinschaftsprojekt zeigen wir, dass die IL9-induzierte Aktivierung von angeborenen lymphoiden Zellen vom Typ 2 (ILC2) eine wichtige Rolle bei der Entzündungsbekämpfung spielt. IL9 induziert die Proliferation von ILC2 und stimuliert deren Potenzial, regulatorische T-Zellen zu aktivieren. Eine Depletion von IL9 oder ILC2 führt zu einer Chronifizierung von Entzündungsprozessen, während rekombinantes IL9 oder der Transfer von ILC2 eine Entzündungshemmung bewirken. Dementsprechend korreliert die Anzahl der ILC2 bei Patienten mit rheumatoider Arthritis umgekehrt mit der klinischen Krankheitsaktivität.
- Palumbo-Zerr K, Zerr P, Distler A, Fliehr J, Mancuso R, Huang J, Mielenz D, Tomcik M, Fürnrohr BG, Scholtysek C, Dees C, Beyer C, Krönke G, Metzger D, Distler O, Schett G, Distler JH. (2015) Orphan nuclear receptor NR4A1 regulates transforming growth factor-β signaling and fibrosis. Nat Med. 21(2):150-8.
In diesem Projekt charakterisieren wir den nukleären Rezeptor NR4A1 als neuen Rückkopplungsmechanismus innerhalb der TGFβ-Signalkaskade. Wir zeigen, dass NR4A1 in physiologischen Gewebereaktionen wie der normalen Wundheilung TGFβ-abhängig induziert wird und dann die Expression von TGFβ-Zielgenen durch Rekrutierung eines Repressorkomplexes hemmt. Bei fibrotischen Erkrankungen wird diese Rückkopplungshemmung durch epigenetische und posttranslationale Mechanismen aufgehoben, NR4A1 wird nicht induziert, und pro-fibrotische Zielgene werden kontinuierlich exprimiert. Die Behandlung mit NR4A1-Agonisten kann diese Rückkopplungshemmung wiederherstellen und zeigt in Mausmodellen für Haut-, Lungen-, Nieren- und Leberfibrose antifibrotische Effekte. Hier identifizieren wir zum ersten Mal die Inaktivierung eines homöostatischen molekularen Rückkopplungshemmungsmechanismus als Ursache der chronischen Gewebereaktion bei fibrotischen Erkrankungen. In Zusammenarbeit mit verschiedenen Pharmaunternehmen arbeiten wir an optimierten NR4A1-Agonisten für den Einsatz in klinischen Studien bei fibrotischen Erkrankungen.
- Dees C, Akhmetshina A, Zerr P, Reich N, Palumbo K, Horn A, Jüngel A, Beyer C, Krönke G, Zwerina J, Reiter R, Alenina N, Maroteaux L, Gay S, Schett G, Distler O, Distler JH. (2011) Platelet-derived serotonin links vascular disease and tissue fibrosis. J Exp Med. 208(5):961-72.
In diesem Projekt wurde zum ersten Mal nachgewiesen, dass Blutplättchen eine wichtige Rolle an der Schnittstelle zwischen Vaskulopathie und fibrotischem Gewebeumbau bei systemischer Sklerose spielen. Die Thrombozyten werden durch die Angiopathie bei systemischer Sklerose zunehmend aktiviert und setzen Serotonin frei. Gleichzeitig wird ein Serotoninrezeptor, 5HTR2B, von Fibroblasten in fibrotischer Haut vermehrt exprimiert. Serotonin aktiviert über 5HTR2B selektiv die Expression von profibrotischen Genen. Mäuse mit einer therapeutischen Hemmung der Thrombozytenaktivierung, einem Mangel an Serotonin in den Thrombozyten oder einer pharmazeutischen oder genetischen Inaktivierung von 5HTR2B sind vor Fibrose geschützt. Diese Arbeit war der Ausgangspunkt für eine erfolgreiche klinische PoC-Studie mit einem 5HTR2B-Antagonisten, eine geplante Phase-3-Studie, und ist die Grundlage für weitere klinische Programme mit hochselektiven 5HTR2B-Inhibitoren bei systemischer Sklerose und anderen fibrosierenden Erkrankungen.
- Distler JH, Jüngel A, Huber LC, Seemayer CA, Reich CF 3rd, Gay RE, Michel BA, Fontana A, Gay S, Pisetsky DS, Distler O. (2005) The induction of matrix metalloproteinase and cytokine expression in synovial fibroblasts stimulated with immune cell microparticles. Proc Natl Acad Sci U S A. 102(8):2892-7.
In früheren Arbeiten hatten wir gezeigt, dass verschiedene Immunzellen nach ihrer Aktivierung oder bei Apoptose Mikropartikel von der Zellmembran ablösen, die als Vesikel mit verschiedenen intrazellulären Komponenten gefüllt sind. In dieser Arbeit zeigen wir, dass Mikropartikel bei Entzündungsprozessen als neuartiger Kommunikationsmechanismus zwischen Immunzellen und mesenchymalen Zellen fungieren. Mikropartikel, die von Immunzellen im Gelenk freigesetzt werden, induzieren einen proinflammatorischen Phänotyp in synovialen Fibroblasten mit Freisetzung von Zytokinen und matrixabbauenden Enzymen. In Folgearbeiten konnten wir außerdem zeigen, dass Mikropartikel auch die Angiogenese durch die Freisetzung angiogener Chemokine und durch Wirkungen auf endotheliale Vorläuferzellen regulieren. Außerdem konnten wir zeigen, dass Mikropartikel im Plasma mit der Aktivität und den klinischen Manifestationen verschiedener rheumatologischer Erkrankungen korrelieren.
PubMed Publikationsliste von Prof. Dr. Jörg Distler